Vollständige Haarlosigkeit Ursache des IFAP-Syndroms
11.06.2009, 15:20 UhrMarburger Mediziner haben die Ursache des so genannten IFAP-Syndroms gefunden, einer seltenen Erbkrankheit, die bei Knaben und Männern zu vollständiger Haarlosigkeit und erhöhter Lichtempfindlichkeit führen kann.
Marburger Mediziner haben die Ursache des so genannten IFAP-Syndroms gefunden, einer seltenen Erbkrankheit, die bei Knaben und Männern zu vollständiger Haarlosigkeit und erhöhter Lichtempfindlichkeit führen kann. Wie die Forscher berichten, beruht das Leiden auf Fehlern in der DNA-Sequenz des zugrunde liegenden Gens.
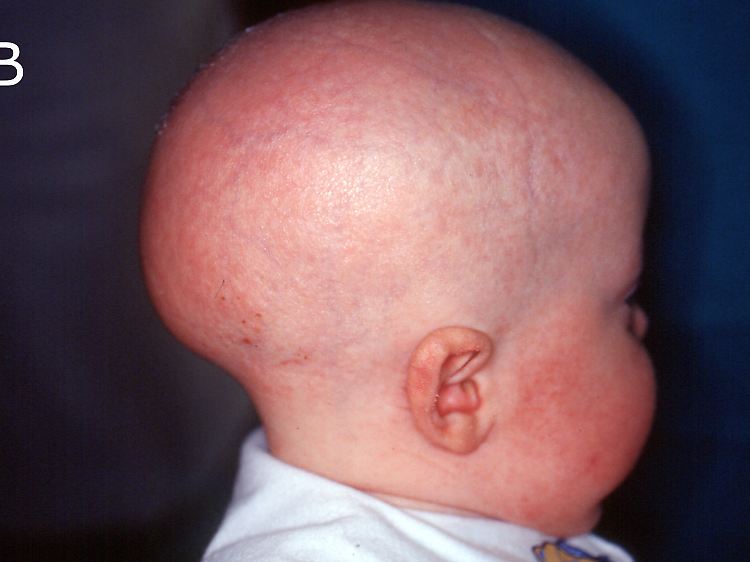
Diesem Jungen fehlt jegliche Behaarung (Atrichie).
Drei Leitsymptome kennzeichnen das so genannte IFAP-Syndrom ("Ichthyosis follicularis mit Atrichie und Photophobie"): Gänsehautartige Verhornungsstörung der Haut, Lichtempfindlichkeit aufgrund von Hornhautschädigungen der Augen und das Fehlen jeglicher Behaarung. Zu diesen Hauptmerkmalen treten manchmal neurologische Störungen, allergische Reaktionen, entzündlicher Darmverschluss sowie Auffälligkeiten an Nieren, Wirbelsäule und Hoden hinzu. Das vollständige Krankheitsbild zeigen nur männliche Patienten. Betroffene Mädchen und Frauen weisen lediglich flecken- oder streifenförmige Areale mit Hautschuppung und erniedrigter Schweißbildung oder vermindertem Haarwuchs auf. Weltweit sind etwa 100 Fälle beschrieben.
Ursache bisher unbekannt
Die Ursache des IFAP-Syndroms, das sich von Familie zu Familie unterschiedlich schwer ausprägt, war bislang unbekannt. Vermutet wurde ein Erbgang, der an das X-Chromosom gebunden ist, von dem Frauen zwei Kopien in jeder Körperzelle tragen, Männer jedoch nur eine. Wie die Wissenschaftler nachweisen konnten, wird die Krankheit durch Mutationen im MBTPS2-Gen verursacht, das tatsächlich auf dem X-Chromosom liegt.
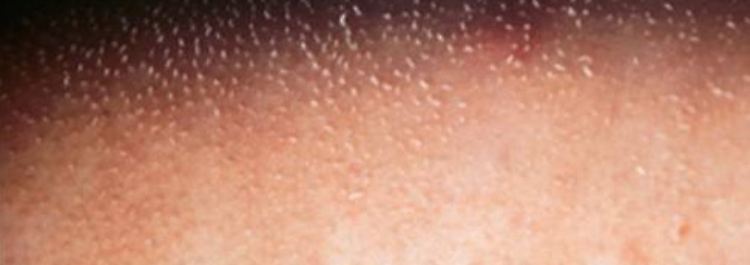
Gänsehautartige Verhornungsstörung der Haut (Ichthyosis).
(Foto: AG Grzeschik)
Diese Erbanlage enthält die Information für ein Enzym, das wichtige Steuerungssignale aktiviert. Die Erkrankung tritt auf, wenn das Genprodukt in seiner Funktion beeinträchtigt ist, weil wichtige Aminosäuren ausgetauscht sind. Hamsterzellen mit dem fehlerhaften Gen können die Funktion des Genprodukts ersetzen, wenn man das funktionstüchtige, unveränderte Enzym von außen einschleust. Je stärker vermindert die Aktivität des Enzyms, desto schwerer ist das Krankheitsbild der männlichen Patienten.
Perspektiven für Betroffene
"Unsere Entdeckung ist von unmittelbar praktischer Bedeutung für die betroffenen Familien", erklärt Senior-Autor Grzeschik. Jetzt könne nicht nur bei männlichen Patienten eine sichere Diagnose gestellt werden, sondern auch bei möglichen Genträgerinnen. In Familien mit ungewöhnlich schwerer Symptomatik lasse sich Klarheit über die genetische Ursache gewinnen. Auch sei mit gewisser Wahrscheinlichkeit eine Vorhersage möglich, wie schwer eine zu erwartende Entwicklungsstörung vermutlich sein wird - je nachdem, in welchem Grade der Austausch eines bestimmten Aminosäurebausteins im MBTPS2-Enzym dessen Funktion beeinträchtigt.
Nicht zuletzt eröffnen die Ergebnisse neue Perspektiven, was die Erforschung der essentiellen Schaltstelle eines lebenswichtigen Entwicklungsprozesses angeht: Ein völliger Ausfall von MBTPS2 ist offenbar mit dem Leben nicht vereinbar. Die Folgen eines teilweisen Verlusts der Enzymfunktion waren bisher unbekannt. Nunmehr lässt sich systematisch ermitteln, welche Störung mit dem Austausch welches Bausteins korreliert ist. Auf diese Weise kann man die Aufgabe einzelner Domänen des Proteins beleuchten.
Quelle: ntv.de, idw